Help
Quick search
You can start a quick search through our main page. Now, we accept 5 kinds of inputs: chromosome position, rs id, gene name, ensembl gene id and disease name, and you can click the demo below the search column. The default reference genome is GRCh38; the default search range for a gene is the position of the gene and its 1kb upstream and downstream region.
Search result
Following is an illustration of the result page of quick search:
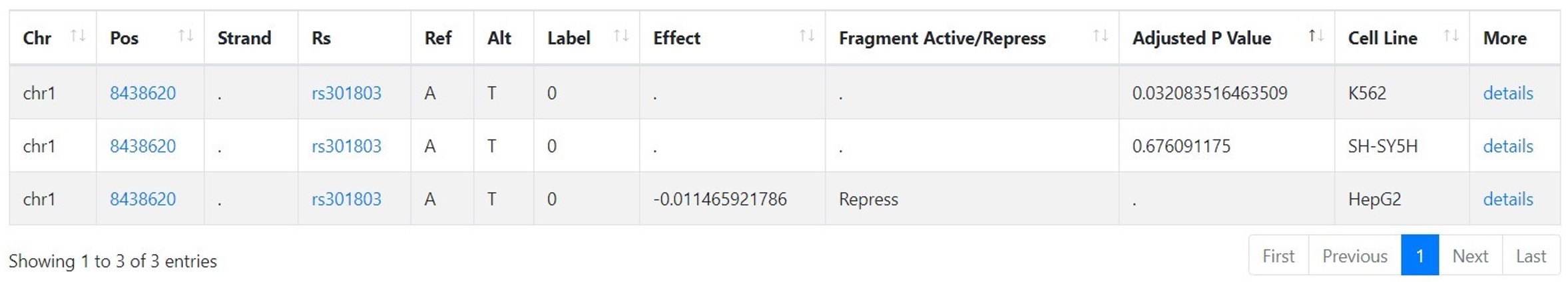
Following columns are presented, in a VCF-based format:
| Column | Note |
|---|---|
| Chr | Chromosome. |
| Pos | Position. The default reference genome is GRCh38. |
| Strand | The direction of the strand. |
| Rs | Reference SNP ID. |
| Ref | Ref allele. |
| Alt | Alt allele. |
| Label | Label "1" is for positive variants, which are considered to have effect on gene expression significantly; otherwise, variants are negative variants and label is "0". The label is given based on adjusted p value and cutoff. |
| Effect | Log2 fold change in expression level. |
| Fragment Active/Repress | The influence of the fragment that contains the variant: "Active", "Repress" or "N.E". This is given based on Effect. If Effect is more than 0, "Active"; if Effect is less than 0, "Repress"; otherwise, "N.E". |
| Adjusted P Value | Adjusted p value. |
| Cell Line | The experimental cell line to test the effect of variants on expression. All variants in REVA are from human. |
| More | Click it for more details |
Variant detail
At the top of variant detail page, you can see the navigation bar:

Baic Information. This section contains the basic information of the variant, including position, rs id, reference and alternative allele,
reference genome version, cell line, label and accession number. You can click position to UCSC genome browser and rs id to
dbSNP. If the column of accession shows ![]() ,
it means that the variant is included in meta-analysis. For these variants, the cutoff of meta p value (presented in the column of adjusted p value)
is 0.001.
,
it means that the variant is included in meta-analysis. For these variants, the cutoff of meta p value (presented in the column of adjusted p value)
is 0.001.
Cell Line and Expression. This section contains all relevent expression information across different cell lines (target cell line is highlighted), including cell line, effect, adjusted p value, label, fragment active/repress, TF and TF active/repress.
| Column | Note |
|---|---|
| Region | The region of the variant on genome. |
| TF | The transcription factor may be influenced by the variant. |
| TF Active/Repress | The influence of the transcription factor on gene expression. "Active" means that the binding of TF can activate gene express; "Repress" means the binding of TF can repress gene expression. For example, the variant may disrupt the binding of repressor TF, so the effect of the variant is activating gene expression (presented in Effect and Fragment Active/Repress). |
Three-dimensional Interacting Gene. The section contains 3D interacting gene inormation from 3DSNP, including gene name, cell type, tissue and loop type. You can click gene name for more information.
Chromatin State. This section contains chromatin state identified by the Core 15-state ChromHMM model through 3DSNP.
Disease and Phenotype. This section contains disease and phenotye information from ClinVar, GWAS, DisGeNET and COSMIC.
| Source | Column | Note |
|---|---|---|
| ClinVar | Interpretation | Clinical significance for the variant. |
| Condition | ClinVar's preferred disease name. | |
| ClinVar | You can click it to ClinVar for more information. | |
| GWAS | Risk Allele | Strongest SNP risk allele. |
| Frequency | Reported risk/effect allele frequency associated with strongest SNP in controls. | |
| Mapped Trait | Mapped experimental factor ontology trait. | |
| P Value | Reported p-value for strongest SNP risk allele. | |
| PubMed | You can click it to original literature for more information. | |
| DisGeNET | Disease Name | UMLS disease Name. You can click it for more information. |
| Disease Semantic Type | UMLS semantic type. | |
| DSI | Disease Specificity Index. It reflects if a variant is associated to several or fewer diseases. The DSI ranges from 0.25 to 1 (higher means less diseases). If the DSI is empty, it implies that the variant is associated only to phenotypes. | |
| DPI | Disease Pleiotropy Index. It reflects if multiple diseases associated to the variant are similar among them (belong to the same MeSH disease class, e.g. Cardiovascular Diseases) or are completely different diseases and belong to different disease classes. The DPI ranges from 0 to 1 (higher means more classes). If the variant has no DPI value, it implies that the variant is associated only to phenotypes, or that the associated diseases do not map to any MeSH classes. | |
| Score | Variant-disease association score. The score ranges from 0 to 1 (higher means more evidences). | |
| NofPmids | Number of support literatures. You can click it for more information. | |
| DisGeNET | You can click it to DisGeNET for more information. | |
| COSMIC | Primary Site | The primary tissue/cancer from which the sample originated. |
| Primary Histology | The histological classification of the sample. | |
| FATHMM MKL Score | FATHMM-MKL non-coding score. The functional scores for individual mutations from FATHMM-MKL are in the form of a single p-value, ranging from 0 to 1. Scores above 0.5 are deleterious, but in order to highlight the most significant data in COSMIC, only scores ≥ 0.7 are classified as 'Pathogenic'. Mutations are classed as 'Neutral' if the score is ≤ 0.5. |
Meta Sources. This section contains source's accession number, label, adjusted p value, effect, region, fragment active/repress, TF and TF active/repress. Only available in variants included in meta-analysis.
Accession. This section contains accession information, including accession number, the times of creation and last update for the accession, information about the assay used in the publication (method type, species of variant, original reference genome version, link to raw data and summary of the assay) and the reference. You can also download the summary of the assay for more information.
Annotation. This section contains chromatin profile, DNA physicochemical properties, evolutionary features and annotation download.
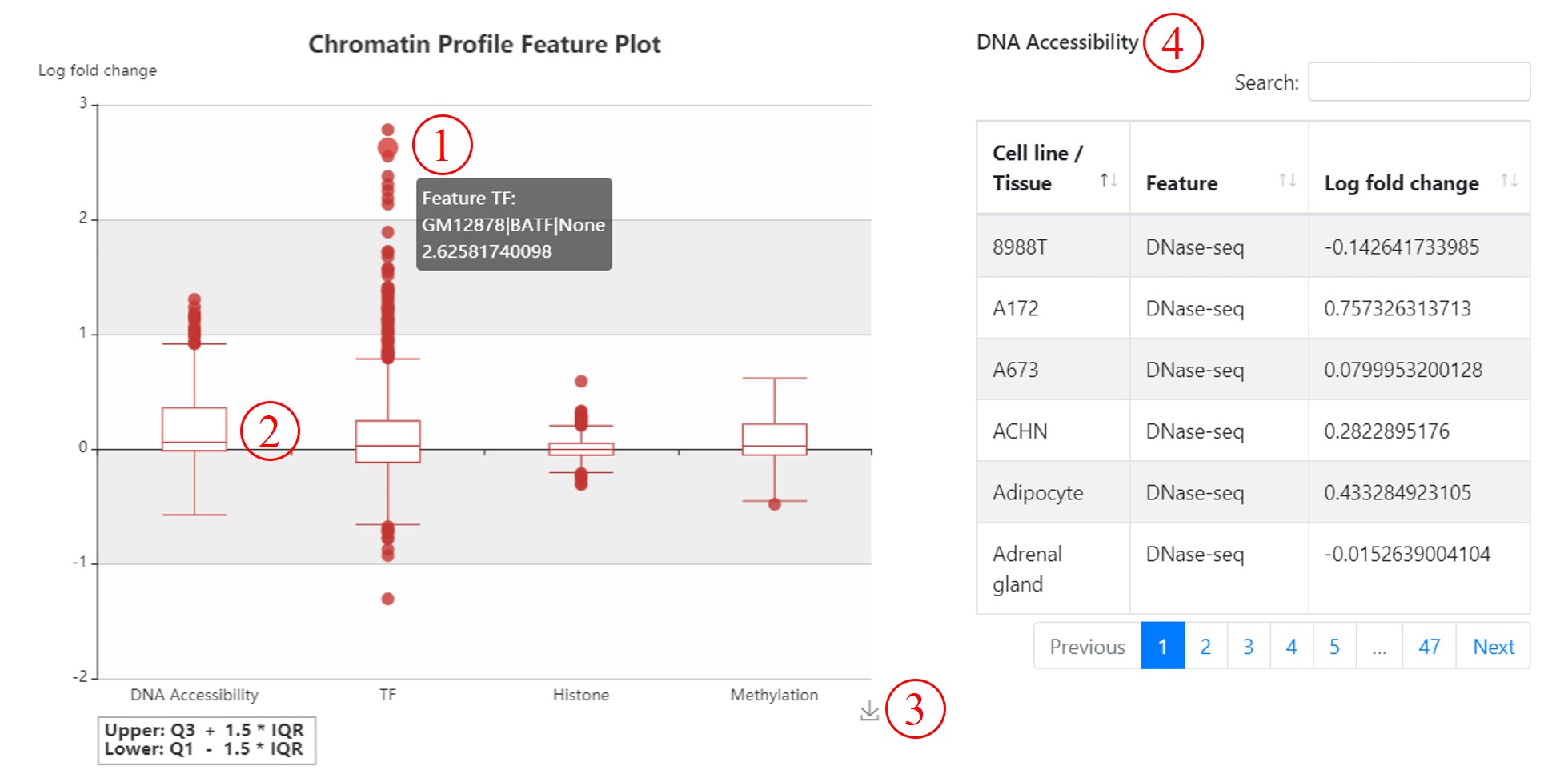
Chromatin Profile Feature Plot. We use a box-plot to show the overall distribution of 2,403 annotations by categoty. You can hover your mouse over the outlier or the box to show more information (1). If you click the box (2), the right table (4) will present the detail information of corresponding category's annotation. The Log fold change is 2 based, if it is more than 0, it means the alternative allele will decrease the binding affinity of related feature (for example, TFs) compring to reference allele; otherwise, increase the binding affinity. You can also save the box-plot as an image to your local computer (3).
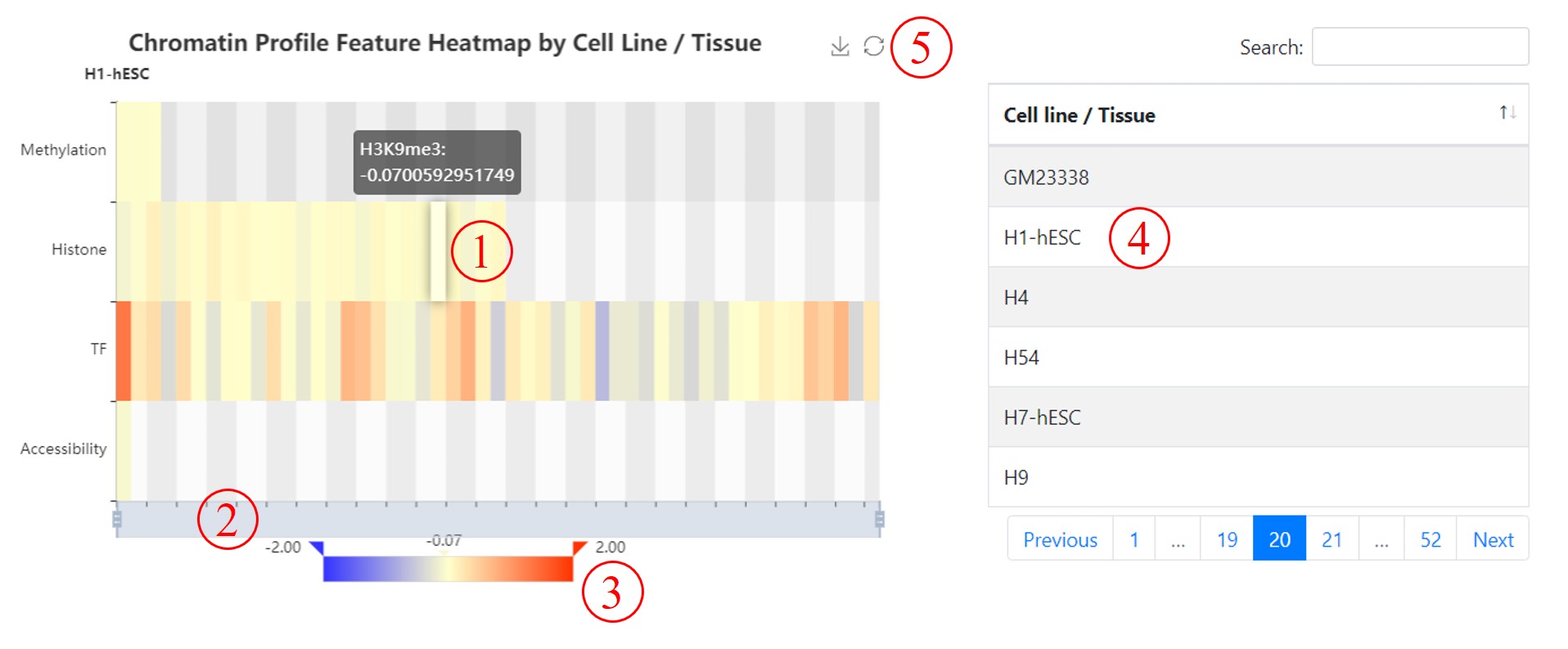
Chromatin Profile Feature Heatmap by Cell Line / Tissue. We also use a heatmap to show annotations by cell line / tissue. By default, it renders the annotations of target cell line of the variant (if the cell line exists in annotations). You can hover your mouse over the block for more information (1). Besides, you can use the slider to foucus on what you are interested in (2) or visualMap to filter annotations (3). You can reset the filter or save the image through (5). Furthermore, you can click the cell line /tissue in right table to render corresponding annotations (4).
DNA Physicochemical Properties and Evolutionary Features. You can hover your mouse over the feature name in DNA physicochemical properties table for more information.
All 2,424 annotations can be downloaded by clicking the button at the end of Annotation section for further analysis.
Advanced search
For customized search, we provide combined search and batch search. You can click here for more information.
Benchmark
Seven state-of-the-art tools which were developed to assess the functional impact of noncoding variants were involved in our benchmark. You can click here for more information.
Download
You can click here to download all variants in REVA and the benchmarking dataset.
Contact us
Team: Yu Wang, Fangyuan Shi, Yu Liang, Ge Gao.
Cite: Wang, Y., Shi, F. Y., Liang, Y., & Gao, G. (2021). REVA as a Well-curated Database for Human Expression-modulating Variants. Genomics, Proteomics & Bioinformatics 19, 590–601.
Mail: [email protected]